|
|
撰写: 米妮 来源:小张聊科研平台的“ i生信”公众号,微信公众号搜索“ i生信”即可关注/扫描关注见文末
#生信分析# #生信发文# #数据库#
文章看得多的小伙伴都知道TCGA数据库,大多研究者在文章里都利用过TCGA的数据来验证他们的数据分析结果,所以TCGA数据的下载和分析,作为一名生信人员也必会,虽然我也下过,但是每次都忘记自己之前怎么下,都得重新研究,所以这次记下来和有需要的小伙伴一起分享~
TCGA (The cancer genome atlas, 癌症基因组图谱),由美国National Cancer Institute(NCI)和National Human Genome Research Institute(NHGRI)于2006年联合启动的项目,目前TCGA已经收录了各种人类癌症包括每个癌症亚型在内的临床数据,基因组,转录组,甲基化等数据,是癌症研究者验证数据的重要数据来源。
官网会由于国内网问题,需要耐心等加载,主页如图1所示:
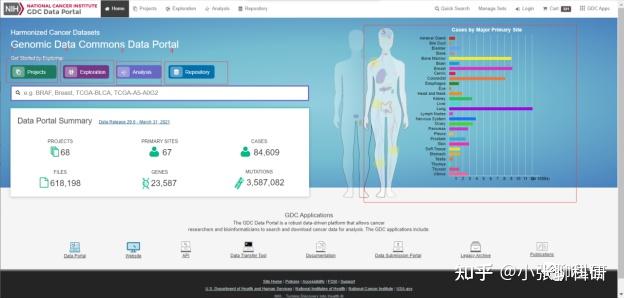
图 1
1.主要按project来寻找数据
2.主要按样本的实验信息和临床信息来找寻数据
3.网页分析功能
4.整合样本临床信息和样本数据信息来筛选数据
5.按样本的组织类型查询数据点击后与2的页面一致
(https://portal.gdc.cancer.gov)
下载manifest 文件
示例带大家一起下载下肾癌的表达矩阵选择标准化后的FPKM表达矩阵,其它类似的数据下载类似,点击‘Repository’进入图2
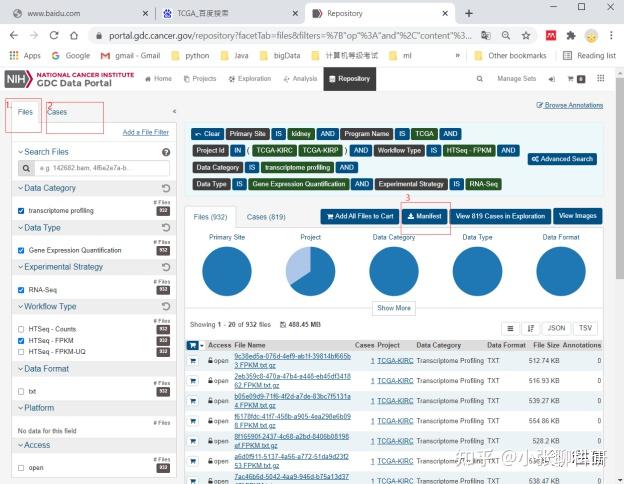
图 2
1.Files选择样本得数据类型
2.选择样本的临床特征
3.下载选择好的manifast,利用此mnifast为后续数据下载做准备,此案例演
示下载kidney的表达矩阵,数据为标准化的FPKM值,样本包括
Kidney cancer的KIRC和KIRP
下载manifest选中样本的元数据
下载manifest 选中样本的原数据,一般包括样本信息和实验测序样本编号的信息,在刚才的选择页面上点击如图3:
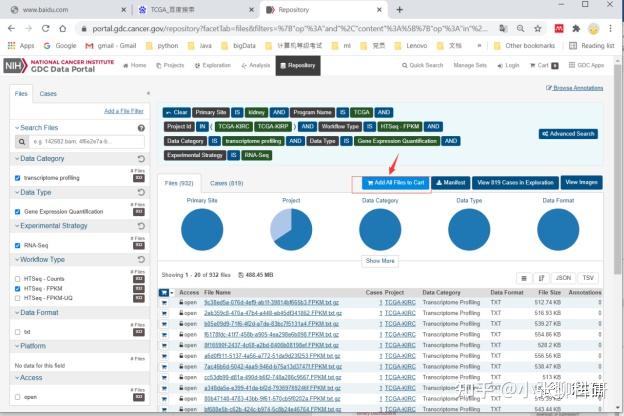
图 3
先把选好的样本添加到 Cart,然后下载metadata,如图4:
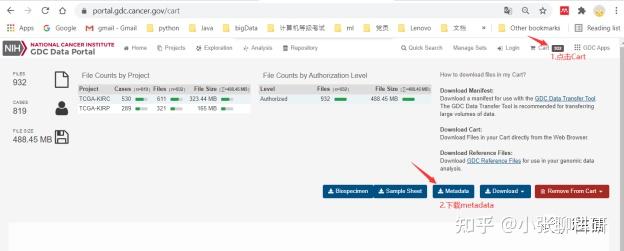
图 4
读入json在R中可以看到:元数据中有它的文件名,如图5:
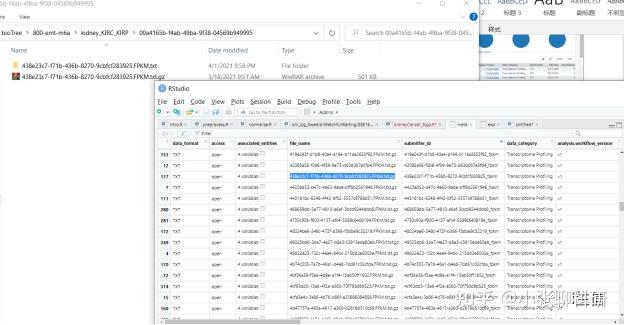
图 5
数据下载工具——gdc-client的下载
数据的下载要利用gdc-client,下载地址:
https://gdc.cancer.gov/access-data/gdc-data-transfer-tool
下载过程看图6:
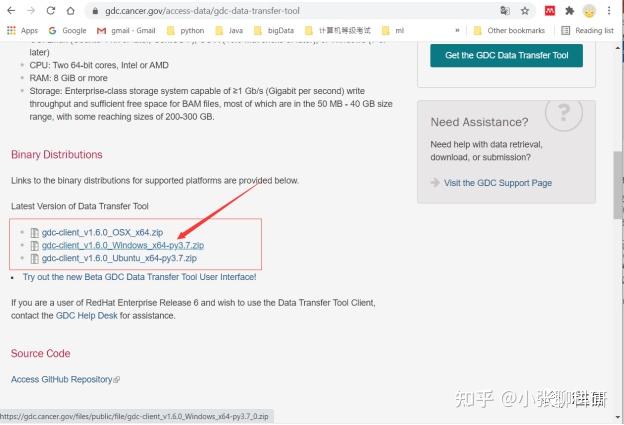
图 6
给大家演示的是在Windows上的,下载后解压,配置环境变量后就可以使用了,如图7&8:
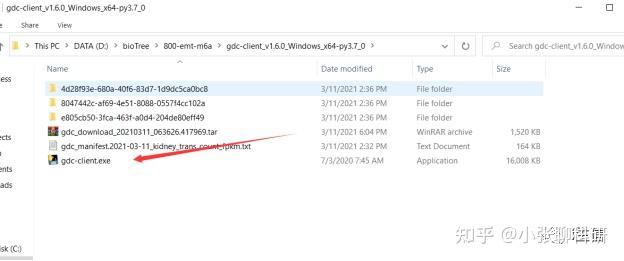
图 7
图 8
现在指定刚才从TCGA下载的 manifest文件开始下载文件,下载在当前路径下,提前进入数据下载目录:
gdc-client download -m gdc_manifest.2021-03-16_kidney.txt

同样也是有时网不好会Connection error,需要多试几次,等数据下载好就可以了~
合并所有样本数据
下载好的文件是长这样的,如图9&10,也就是一个样本在一个文件夹,一个文件夹有一个txt文件,二列数据,整理起来要是不用代码,得发疯,下面利用代码R,就可以自动合并样本得到所有样本的表达矩阵。
图 9
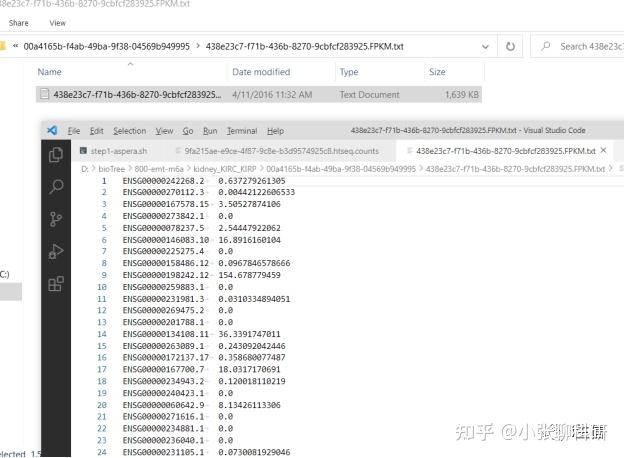
图 10
利用代码实现自动读取每个文件夹下的下载数据:
setwd('D:/bioTree/800-emt-m6a/')
# count_files = dir("kidney_fpkm/",pattern = "*.htseq.counts.gz$",recursive = T)
# 1. kidney cancer 表达数据的整理
fpkm_files = dir("kidney_KIRC_KIRP/",pattern = "*.FPKM.txt.gz$",recursive = T)
ex = function(x){
result <- read.table(file.path(&#34;kidney_KIRC_KIRP/&#34;,x),row.names = 1,sep = &#34;\t&#34;)
return(result)
}
exp = lapply(fpkm_files,ex)
exp <- do.call(cbind,exp)
dim(exp)
把样本数据对应到元数据中的样本名
上一步得到的exp是没有样本列名,需要读入meta信息,利用样本合并的文件顺序来到元数据中匹配样本名,把样本ID重新作为列名。
meta <- jsonlite::fromJSON(&#34;metadata.cart.2021-03-18_KIRP_FPKM.json&#34;)
colnames(meta)
ids <- meta$associated_entities;class(ids)
ids[[1]]
class(ids[[1]][,1])
ID = sapply(ids,function(x){x[,1]}) ## 文件id
file2id = data.frame(file_name = meta$file_name,
ID = ID)
head(file2id$file_name)
head(fpkm_files)
fpkm_files2 = stringr::str_split(fpkm_files,&#34;/&#34;,simplify = T)[,2]
fpkm_files2[1] %in% file2id$file_name
file2id = file2id[match(fpkm_files2,file2id$file_name),]
colnames(exp) = file2id$ID
exp[1:4,1:4]
dim(exp)
得到的矩阵如下:
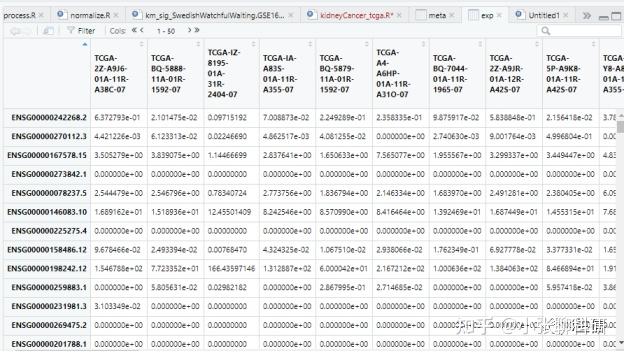
根据样本ID区分样本是癌症还是对照
Barcode含义介绍:
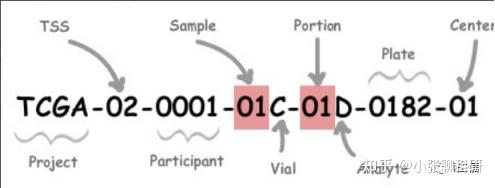
详细介绍参见官网:
https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/
看样本类型主要看Sample类的编码:
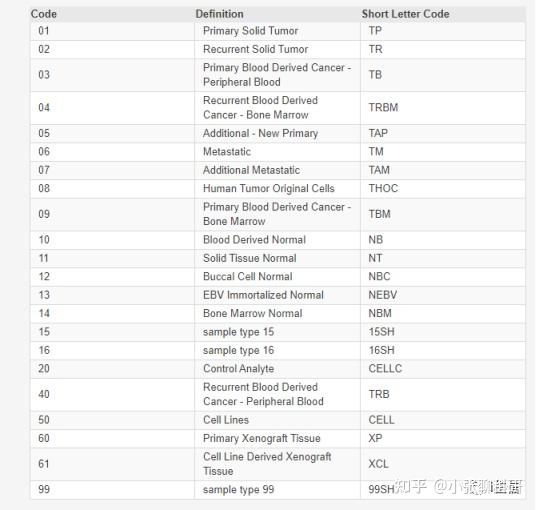
接下来对表达矩阵进行样本组的区分就可以进行下游分析了,不花钱的验证数据就到手了~
原文链接: |
|



 发表于 2023-3-1 17:40:48
发表于 2023-3-1 17:40:48